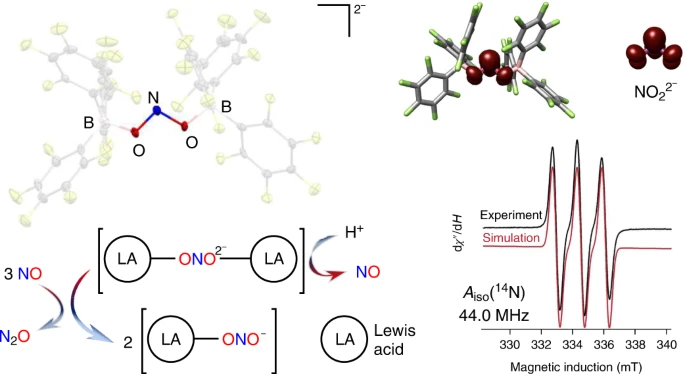
ABSTRACT: Reduction of nitrite anions (NO2–) to nitric oxide (NO), nitrous oxide (N2O) and ultimately dinitrogen (N2) takes place in a variety of environments, including in the soil as part of the biogeochemical nitrogen cycle and in acidified nuclear waste. Nitrite reduction typically takes place within the coordination sphere of a redox-active transition metal. Here we show that Lewis acid coordination can substantially modify the reduction potential of this polyoxoanion to allow for its reduction under non-aqueous conditions (–0.74 V versus NHE). Detailed characterization confirms the formation of the borane-capped radical nitrite dianion (NO22–), which features a N(II) oxidation state. Protonation of the nitrite dianion results in the facile loss of nitric oxide (NO), whereas its reaction with NO results in disproportionation to nitrous oxide (N2O) and nitrite (NO2–). This system connects three redox levels in the global nitrogen cycle and provides fundamental insights into the conversion of NO2– to NO.
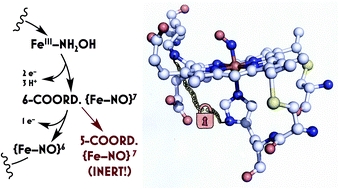
ABSTRACT: Ammonia (NH3)-oxidizing bacteria (AOB) derive total energy for life from the multi-electron oxidation of NH3 to nitrite (NO2−). One obligate intermediate of this metabolism is hydroxylamine (NH2OH), which can be oxidized to the potent greenhouse agent nitrous oxide (N2O) by the AOB enzyme cytochrome (cyt) P460. We have now spectroscopically characterized a 6-coordinate (6c) {FeNO}7 intermediate on the NH2OH oxidation pathway of cyt P460. This species has two fates: it can either be oxidized to the {FeNO}6 that then undergoes attack by NH2OH to ultimately generate N2O, or it can lose its axial His ligand, thus generating a stable, off-pathway 5-coordinate (5c) {FeNO}7 species. We show that the wild type (WT) cyt P460 exhibits a slow nitric oxide (NO)-independent conversion (kHis-off = 2.90 × 10−3 s−1), whereas a cross-link-deficient Lys70Tyr cyt P460 mutant protein underwent His dissociation via both a NO-independent (kHis-off = 3.8 × 10−4 s−1) and a NO-dependent pathway [kHis-off(NO) = 790 M−1 s−1]. Eyring analyses of the NO-independent pathways for these two proteins revealed a significantly larger (ca. 27 cal mol−1 K−1) activation entropy (ΔS‡) in the cross-link-deficient mutant. Our results suggest that the Lys–heme cross-link confers rigidity to the positioning of the heme P460 cofactor to avoid the fast NO-dependent His dissociation pathway and subsequent formation of the off-pathway 5c {FeNO}7 species. The relevance of these findings to NO signaling proteins such as heme-nitric oxide/oxygen binding (H-NOX) is also discussed.
.svg)