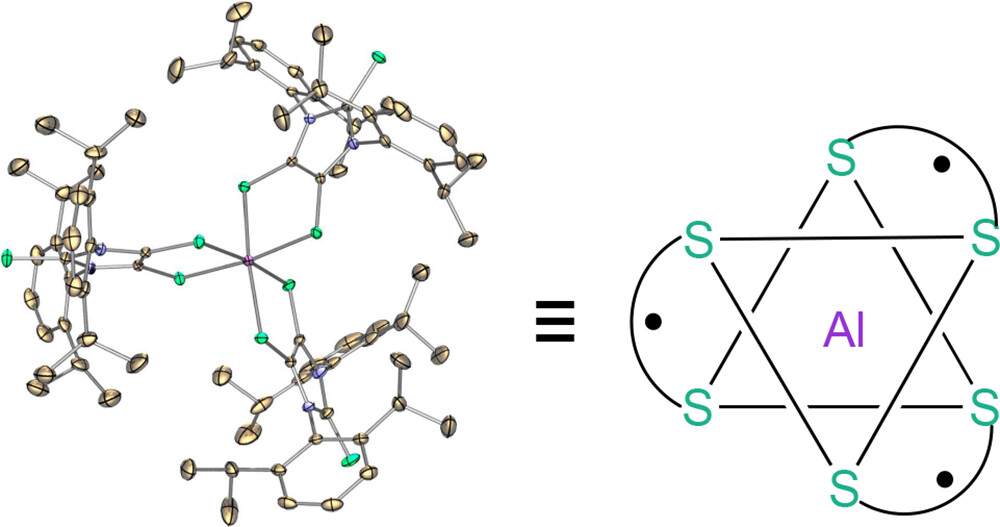
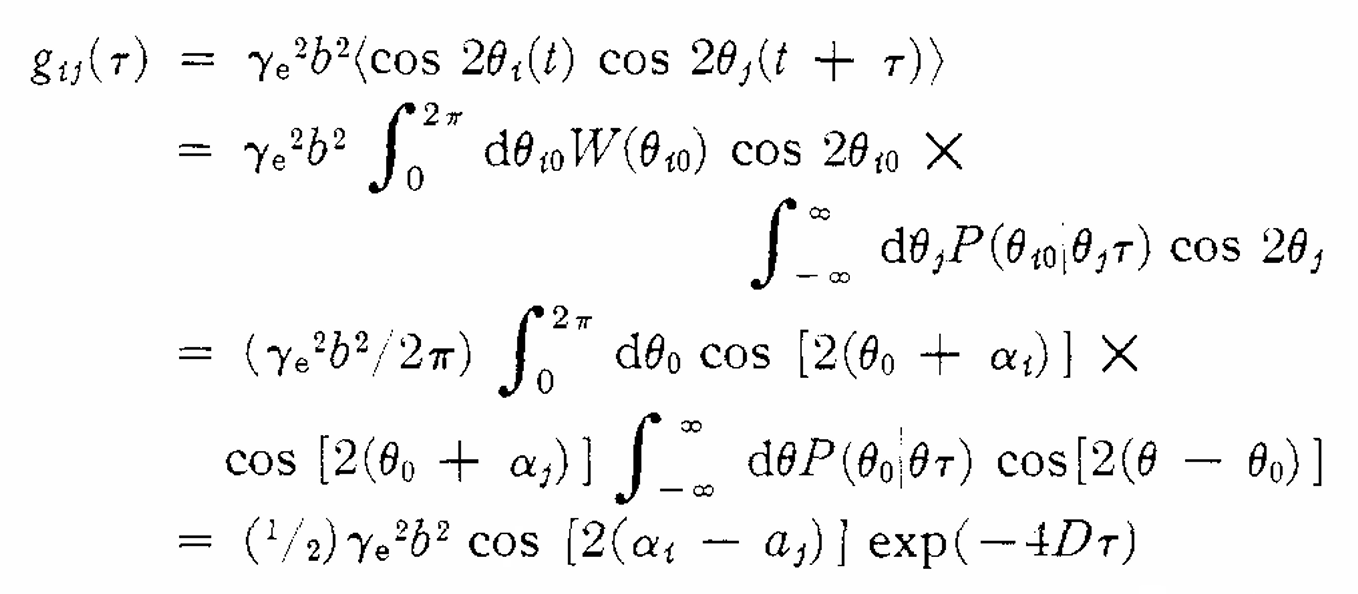ABSTRACT: A stable aluminum tris(dithiolene) triradical (3) was experimentally realized through a low-temperature reaction of the sterically demanding lithium dithiolene radical (2) with aluminum iodide. Compound 3 was characterized by single-crystal X-ray diffraction, UV–vis and EPR spectroscopy, SQUID magnetometry, and theoretical computations. The quartet ground state of triradical 3 has been unambiguously confirmed by variable-temperature continuous wave EPR experiments and SQUID magnetometry. Both SQUID magnetometry and broken-symmetry DFT computations reveal a small doublet–quartet energy gap [ΔEDQ = 0.18 kcal mol–1 (SQUID); ΔEDQ = 0.14 kcal mol–1 (DFT)]. The pulsed EPR experiment (electron spin echo envelop modulation) provides further evidence for the interaction of these dithiolene-based radicals with the central aluminum nucleus of 3.
ABSTRACT: Hydrogen-atom transfer mediated by earth-abundant transition-metal hydrides (M-Hs) has emerged as a powerful tool in organic synthesis. Current methods to generate M-Hs most frequently rely on oxidatively initiated hydride transfer. Herein, we report a reductive approach to generate Co–H, which allows for canonical hydrogen evolution reactions to be intercepted by hydrogen-atom transfer to an alkene. Electroanalytical and spectroscopic studies provided mechanistic insights into the formation and reactivity of Co–H, which enabled the development of two new alkene hydrofunctionalization reactions.
ABSTRACT: Distance distribution information obtained by pulsed dipolar EPR spectroscopy provides an important contribution to many studies in structural biology. Increasingly, such information is used in integrative structural modeling, where it delivers unique restraints on the width of conformational ensembles. In order to ensure reliability of the structural models and of biological conclusions, we herein define quality standards for sample preparation and characterization, for measurements of distributed dipole–dipole couplings between paramagnetic labels, for conversion of the primary time‐domain data into distance distributions, for interpreting these distributions, and for reporting results. These guidelines are substantiated by a multi‐laboratory benchmark study and by analysis of data sets with known distance distribution ground truth. The study and the guidelines focus on proteins labeled with nitroxides and on double electron–electron resonance (DEER aka PELDOR) measurements and provide suggestions on how to proceed analogously in other cases.
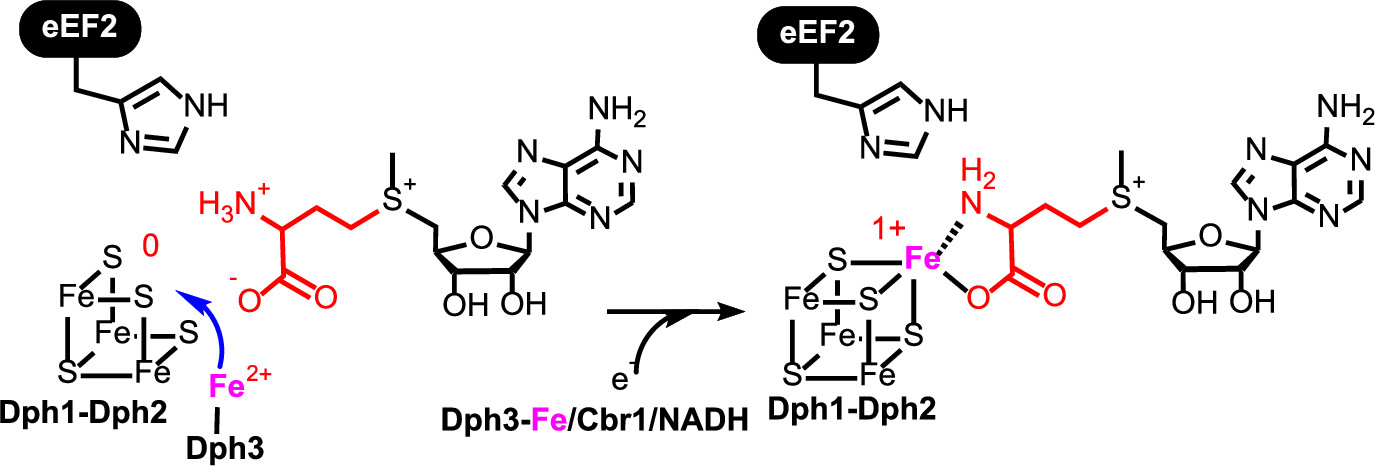
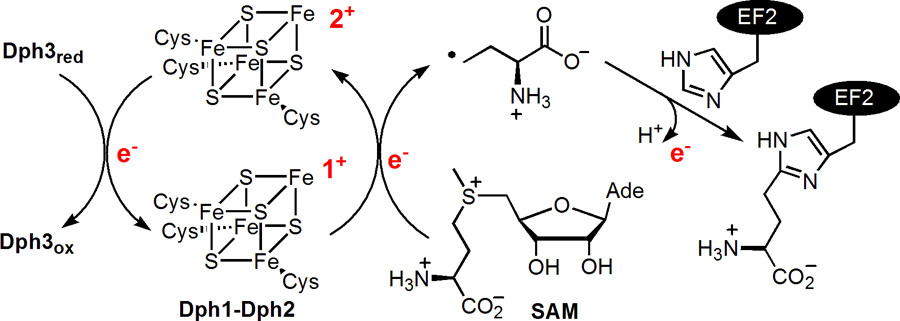
ABSTRACT: All radical S‐adenosylmethionine (radical‐SAM) enzymes, including the noncanonical radical‐SAM enzyme diphthamide biosynthetic enzyme Dph1–Dph2, require at least one [4Fe–4S](Cys)3 cluster for activity. It is well‐known in the radical‐SAM enzyme community that the [4Fe–4S](Cys)3 cluster is extremely air‐sensitive and requires strict anaerobic conditions to reconstitute activity in vitro. Thus, how such enzymes function in vivo in the presence of oxygen in aerobic organisms is an interesting question. Working on yeast Dph1–Dph2, we found that consistent with the known oxygen sensitivity, the [4Fe–4S] cluster is easily degraded into a [3Fe–4S] cluster. Remarkably, the small iron‐containing protein Dph3 donates one Fe atom to convert the [3Fe–4S] cluster in Dph1–Dph2 to a functional [4Fe–4S] cluster during the radical‐SAM enzyme catalytic cycle. This mechanism to maintain radical‐SAM enzyme activity in aerobic environments is likely general, and Dph3‐like proteins may exist to keep other radical‐SAM enzymes functional in aerobic environments.
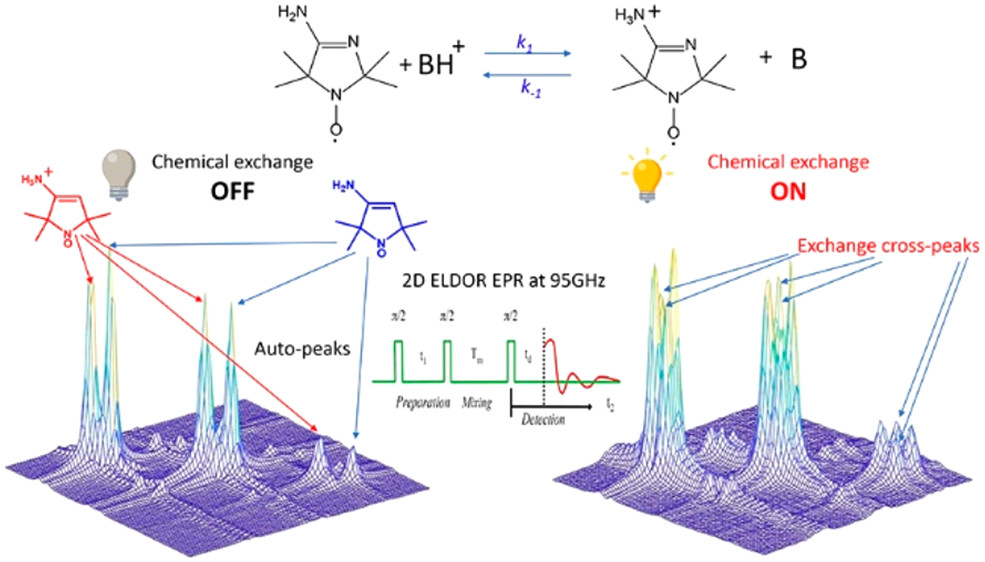
ABSTRACT: Exchange processes which include conformational change, protonation/deprotonation, and binding equilibria are routinely studied by 2D exchange NMR techniques, where information about the exchange of nuclei between environments with different NMR shifts is obtained from the development of cross‐peaks. Whereas 2D NMR enables the real time study of millisecond and slower exchange processes, 2D ESR in the form of 2D‐ELDOR (two‐dimensional electron‐electron double resonance) has the potential for such studies over the nanosecond to microsecond real time scales. Cross‐peak development due to chemical exchange has been seen previously for semiquinones in ESR, but this is not possible for most common ESR probes, such as nitroxides, studied at typical ESR frequencies because, unlike NMR, the exchanging states yield ESR signals that are not resolved from each other within their respective line widths. But at 95 GHz, it becomes possible to resolve them in many cases because of the increased g‐factor resolution. The 95 GHz instrumental developments occurring at ACERT now enable such studies. We demonstrate these new capabilities in two studies: (A) the protonation/deprotonation process for a pH‐sensitive imidazoline spin label in aqueous solution where the exchange rate and the population ratio of the exchanging states are controlled by the concentration and pH of the buffer solution, respectively, and (B) a nitroxide radical partitioning between polar (aqueous) and nonpolar (phospholipid) environments in multilamellar lipid vesicles, where the cross‐peak development arises from the exchange of the nitroxide between the two phases. This work represents the first example of the observation and analysis of cross‐peaks arising from chemical exchange processes involving nitroxide spin labels.
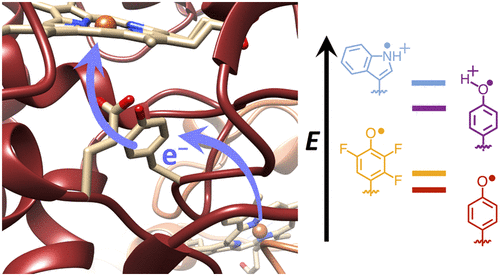
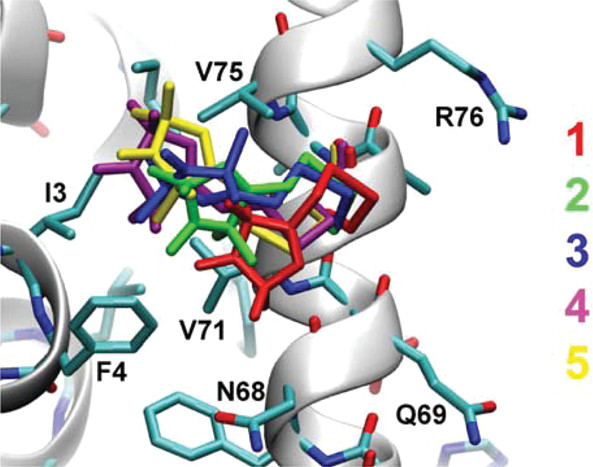
ABSTRACT: Transient tyrosine and tryptophan radicals play key roles in the electron transfer (ET) reactions of photosystem (PS) II, ribonucleotide reductase (RNR), photolyase, and many other proteins. However, Tyr and Trp are not functionally interchangeable, and the factors controlling their reactivity are often unclear. Cytochrome c peroxidase (CcP) employs a Trp191⋅+ radical to oxidize reduced cytochrome c (Cc). Although a Tyr191 replacement also forms a stable radical, it does not support rapid ET from Cc. Here we probe the redox properties of CcP Y191 by non-natural amino acid substitution, altering the ET driving force and manipulating the protic environment of Y191. Higher potential fluorotyrosine residues increase ET rates marginally, but only addition of a hydrogen bond donor to Tyr191⋅ (via Leu232His or Glu) substantially alters activity by increasing the ET rate by nearly 30-fold. ESR and ESEEM spectroscopies, crystallography, and pH-dependent ET kinetics provide strong evidence for hydrogen bond formation to Y191⋅ by His232/Glu232. Rate measurements and rapid freeze quench ESR spectroscopy further reveal differences in radical propagation and Cc oxidation that support an increased Y191⋅ formal potential of ∼200 mV in the presence of E232. Hence, Y191 inactivity results from a potential drop owing to Y191⋅+ deprotonation. Incorporation of a well-positioned base to accept and donate back a hydrogen bond upshifts the Tyr⋅ potential into a range where it can effectively oxidize Cc. These findings have implications for the YZ/YD radicals of PS II, hole-hopping in RNR and cryptochrome, and engineering proteins for long-range ET reactions.
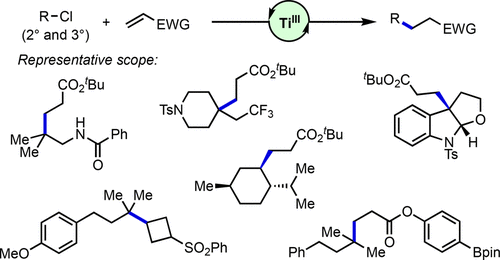
ABSTRACT: Alkyl chlorides are common functional groups in synthetic organic chemistry. However, the engagement of unactivated alkyl chlorides, especially tertiary alkyl chlorides, in transition-metal-catalyzed C–C bond formation remains challenging. Herein, we describe the development of a TiIII-catalyzed radical addition of 2° and 3° alkyl chlorides to electron-deficient alkenes. Mechanistic data are consistent with inner-sphere activation of the C–Cl bond featuring TiIII-mediated Cl atom abstraction. Evidence suggests that the active TiIII catalyst is generated from the TiIV precursor in a Lewis-acid-assisted electron transfer process.
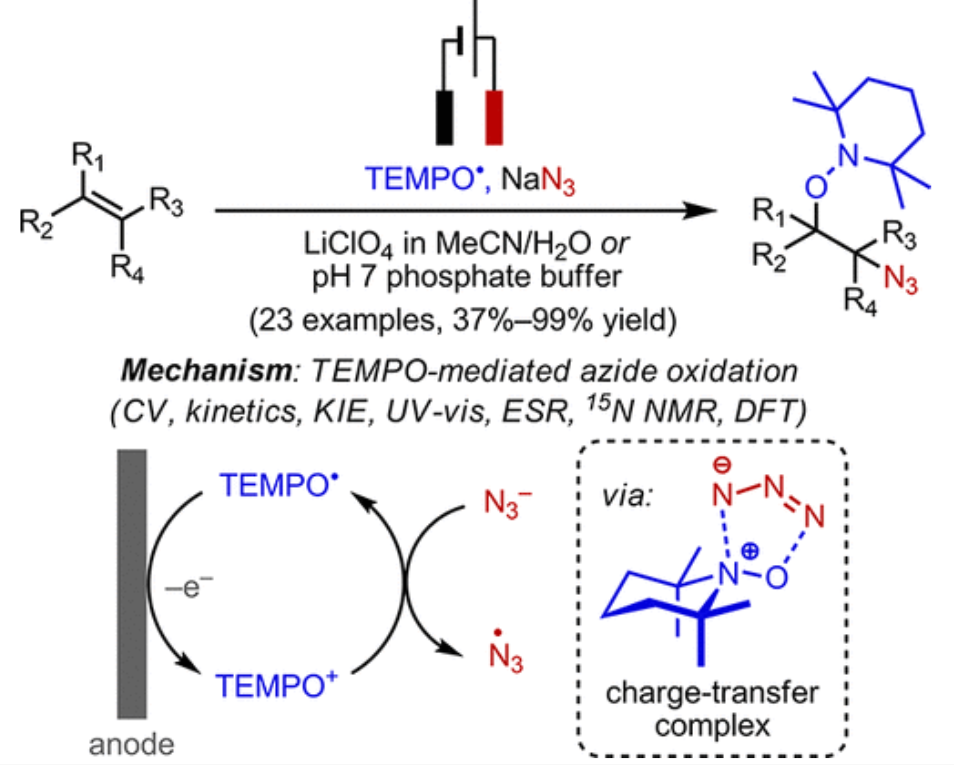
ABSTRACT: We report a mild and efficient electrochemical protocol to access a variety of vicinally C–O and C–N difunctionalized compounds from simple alkenes. Detailed mechanistic studies revealed a distinct reaction pathway from those previously reported for TEMPO-mediated reactions. In this mechanism, electrochemically generated oxoammonium ion facilitates the formation of azidyl radical via a charge-transfer complex with azide, TEMPO–N3. DFT calculations together with spectroscopic characterization provided a tentative structural assignment of this charge-transfer complex. Kinetic and kinetic isotopic effect studies revealed that reversible dissociation of TEMPO–N3 into TEMPO⋅ and azidyl precedes the addition of these radicals across the alkene in the rate-determining step. The resulting azidooxygenated product could then be easily manipulated for further synthetic elaborations. The discovery of this new reaction pathway mediated by the TEMPO+/TEMPO⋅ redox couple may expand the scope of aminoxyl radical chemistry in synthetic contexts.
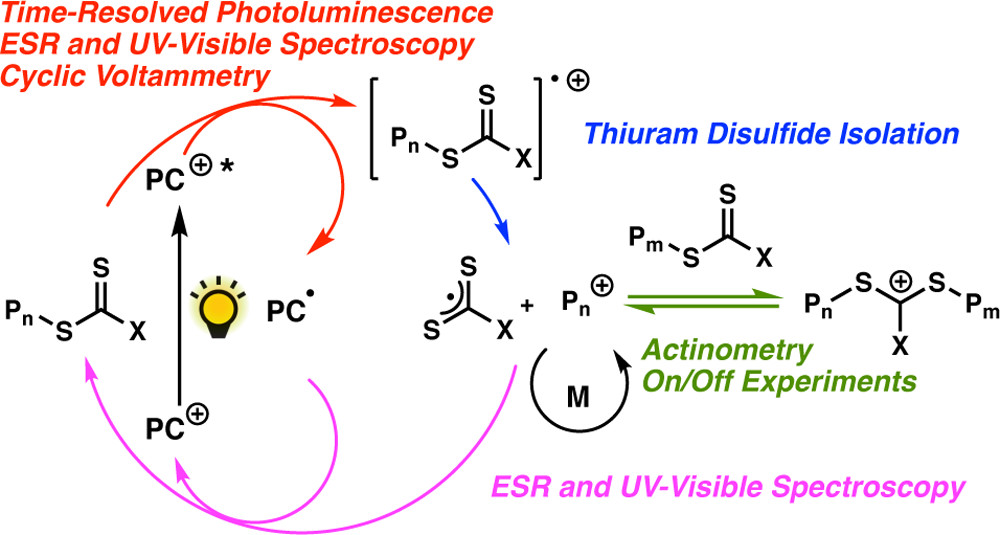
ABSTRACT: The mechanism of the recently reported photocontrolled cationic polymerization of vinyl ethers was investigated using a variety of catalysts and chain-transfer agents (CTAs) as well as diverse spectroscopic and electrochemical analytical techniques. Our study revealed a complex activation step characterized by one-electron oxidation of the CTA. This oxidation is followed by mesolytic cleavage of the resulting radical cation species, which leads to the generation of a reactive cation–this species initiates the polymerization of the vinyl ether monomer–and a dithiocarbamate radical that is likely in equilibrium with the corresponding thiuram disulfide dimer. Reversible addition–fragmentation type degenerative chain transfer contributes to the narrow dispersities and control over chain growth observed under these conditions. Finally, the deactivation step is contingent upon the oxidation of the reduced photocatalyst by the dithiocarbamate radical concomitant with the production of a dithiocarbamate anion that caps the polymer chain end. The fine-tuning of the electronic properties and redox potentials of the photocatalyst in both the excited and the ground states is necessary to obtain a photocontrolled system rather than simply a photoinitiated system. The elucidation of the elementary steps of this process will aid the design of new catalytic systems and their real-world applications.
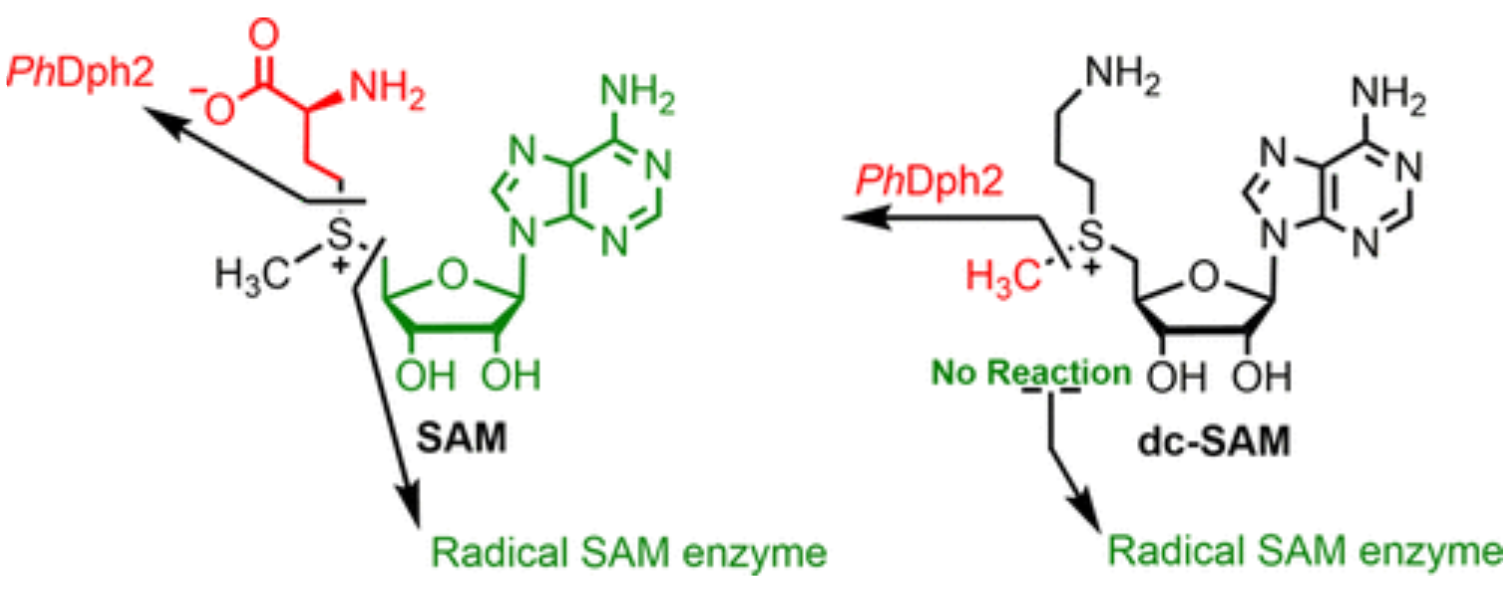
ABSTRACT: S-Adenosylmethionine (SAM) has a sulfonium ion with three distinct C-S bonds. Conventional radical SAM enzymes use a [4Fe-4S] cluster to cleave homolytically the C5′,adenosine-S bond of SAM to generate a 5′-deoxyadenosyl radical, which catalyzes various downstream chemical reactions. Radical SAM enzymes involved in diphthamide biosynthesis, such as Pyrococcus horikoshii Dph2 (PhDph2) and yeast Dph1-Dph2 instead cleave the Cγ,Met-S bond of methionine to generate a 3-amino-3-carboxylpropyl radical. We here show radical SAM enzymes can be tuned to cleave the third C-S bond to the sulfonium sulfur by changing the structure of SAM. With a decarboxyl SAM analogue (dc-SAM), PhDph2 cleaves the Cmethyl-S bond, forming 5′-deoxy-5′-(3-aminopropylthio) adenosine (dAPTA, 1). The methyl cleavage activity, like the cleavage of the other two C-S bonds, is dependent on the presence of a [4Fe-4S]+ cluster. Electron-nuclear double resonance and mass spectroscopy data suggests that mechanistically one of the S atoms in the [4Fe-4S] cluster captures the methyl group from dc-SAM, forming a distinct EPR-active intermediate, which can transfer the methyl group to nucleophiles such as dithiothreitol. This reveals the [4Fe-4S] cluster in a radical SAM enzyme can be tuned to cleave any one of the three bonds to the sulfonium sulfur of SAM or analogues, and is the first demonstration a radical SAM enzyme could switch from an Fe-based one electron transfer reaction to a S-based two electron transfer reaction in a substrate-dependent manner. This study provides an illustration of the versatile reactivity of Fe-S clusters.
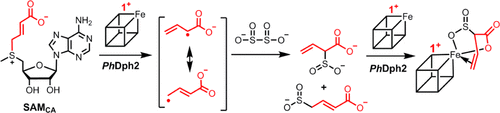
ABSTRACT: Pyrococcus horikoshii Dph2 (PhDph2) is an unusual radical S-adenosylmethionine (SAM) enzyme involved in the first step of diphthamide biosynthesis. It catalyzes the reaction by cleaving SAM to generate a 3-amino-3-carboxypropyl (ACP) radical. To probe the reaction mechanism, we synthesized a SAM analogue (SAMCA), in which the ACP group of SAM is replaced with a 3-carboxyallyl group. SAMCA is cleaved by PhDph2, yielding a paramagnetic (S = 1/2) species, which is assigned to a complex formed between the reaction product, α-sulfinyl-3-butenoic acid, and the [4Fe-4S] cluster. Electron–nuclear double resonance (ENDOR) measurements with 13C and 2H isotopically labeled SAMCA support a π-complex between the C=C double bond of α-sulfinyl-3-butenoic acid and the unique iron of the [4Fe-4S] cluster. This is the first example of a radical SAM-related [4Fe-4S]+ cluster forming an organometallic complex with an alkene, shedding additional light on the mechanism of PhDph2 and expanding our current notions for the reactivity of [4Fe-4S] clusters in radical SAM enzymes.
ABSTRACT: Diphthamide, the target of diphtheria toxin, is a unique posttranslational modification on translation elongation factor 2 (EF2) in archaea and eukaryotes. The biosynthesis of diphthamide was proposed to involve three steps. The first step is the transfer of the 3-amino-3-carboxypropyl group from S-adenosyl-L-methionine (SAM) to the histidine residue of EF2, forming a C–C bond. Previous genetic studies showed this step requires four proteins in eukaryotes, Dph1–Dph4. However, the exact molecular functions for the four proteins are unknown. Previous study showed that Pyrococcus horikoshii Dph2 (PhDph2), a novel iron-sulfur cluster-containing enzyme, forms a homodimer and is sufficient for the first step of diphthamide biosynthesis in vitro. Here we demonstrate by in vitro reconstitution that yeast Dph1 and Dph2 form a complex (Dph1-Dph2) that is equivalent to the homodimer of PhDph2 and is sufficient to catalyze the first step in vitro in the presence of dithionite as the reductant. We further demonstrate that yeast Dph3 (also known as KTI11), a CSL-type zinc finger protein, can bind iron and in the reduced state can serve as an electron donor to reduce the Fe-S cluster in Dph1-Dph2. Our study thus firmly establishes the functions for three of the proteins involved in eukaryotic diphthamide biosynthesis. For most radical SAM enzymes in bacteria, flavodoxins and flavodoxin reductases are believed to serve as electron donors for the Fe-S clusters. The finding that Dph3 is an electron donor for the Fe-S clusters in Dph1-Dph2 is thus interesting and opens up new avenues of research on electron transfer to Fe-S proteins in eukaryotic cells.
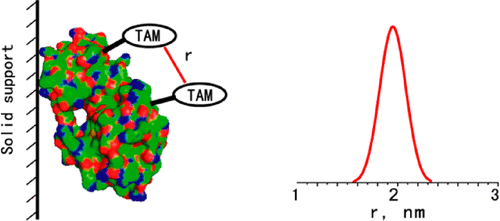
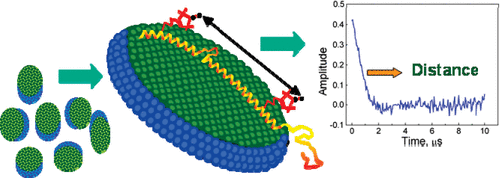
ABSTRACT: Pulsed electron spin resonance (ESR) dipolar spectroscopy (PDS) in combination with site-directed spin labeling is unique in providing nanometer-range distances and distributions in biological systems. To date, most of the pulsed ESR techniques require frozen solutions at cryogenic temperatures to reduce the rapid electron spin relaxation rate and to prevent averaging of electron–electron dipolar interaction due to the rapid molecular tumbling. To enable measurements in liquid solution, we are exploring a triarylmethyl (TAM)-based spin label with a relatively long relaxation time where the protein is immobilized by attachment to a solid support. In this preliminary study, TAM radicals were attached via disulfide linkages to substituted cysteine residues at positions 65 and 80 or 65 and 76 in T4 lysozyme immobilized on Sepharose. Interspin distances determined using double quantum coherence (DQC) in solution are close to those expected from models, and the narrow distance distribution in each case indicates that the TAM-based spin label is relatively localized.
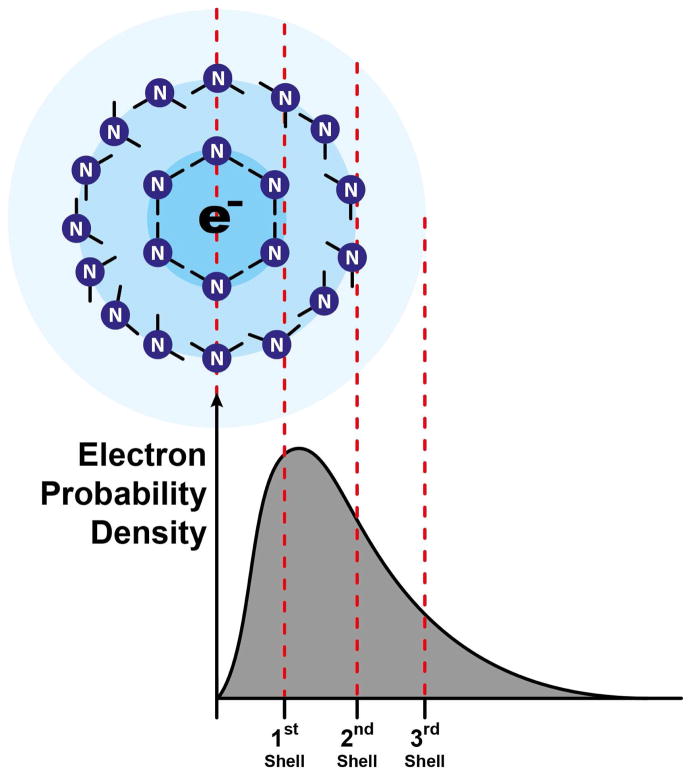
ABSTRACT: Electron transfer or quantum tunneling dynamics for excess or solvated electrons in dilute lithium–ammonia solutions have been studied by pulse electron paramagnetic resonance (EPR) spectroscopy at both X- (9.7 GHz) and W-band (94 GHz) frequencies. The electron spin–lattice (T1) and spin–spin (T2) relaxation data indicate an extremely fast transfer or quantum tunneling rate of the solvated electron in these solutions which serves to modulate the hyperfine (Fermi-contact) interaction with nitrogen nuclei in the solvation shells of ammonia molecules surrounding the localized, solvated electron. The donor and acceptor states of the solvated electron in these solutions are the initial and final electron solvation sites found before, and after, the transfer or tunneling process. To interpret and model our electron spin relaxation data from the two observation EPR frequencies requires a consideration of a multiexponential correlation function. The electron transfer or tunneling process that we monitor through the correlation time of the nitrogen Fermi-contact interaction has a time scale of (1–10) × 10–12 s over a temperature range 230–290 K in our most dilute solution of lithium in ammonia. Two types of electron–solvent interaction mechanisms are proposed to account for our experimental findings. The dominant electron spin relaxation mechanism results from an electron tunneling process characterized by a variable donor–acceptor distance or range (consistent with such a rapidly fluctuating liquid structure) in which the solvent shell that ultimately accepts the transferring electron is formed from random, thermal fluctuations of the liquid structure in, and around, a natural hole or Bjerrum-like defect vacancy in the liquid. Following transfer and capture of the tunneling electron, further solvent-cage relaxation with a time scale of ˜10–13 s results in a minor contribution to the electron spin relaxation times. This investigation illustrates the great potential of multifrequency EPR measurements to interrogate the microscopic nature and dynamics of ultrafast electron transfer or quantum-tunneling processes in liquids. Our results also impact on the universal issue of the role of a host solvent (or host matrix, e.g. a semiconductor) in mediating long-range electron transfer processes and we discuss the implications of our results with a range of other materials and systems exhibiting the phenomenon of electron transfer.
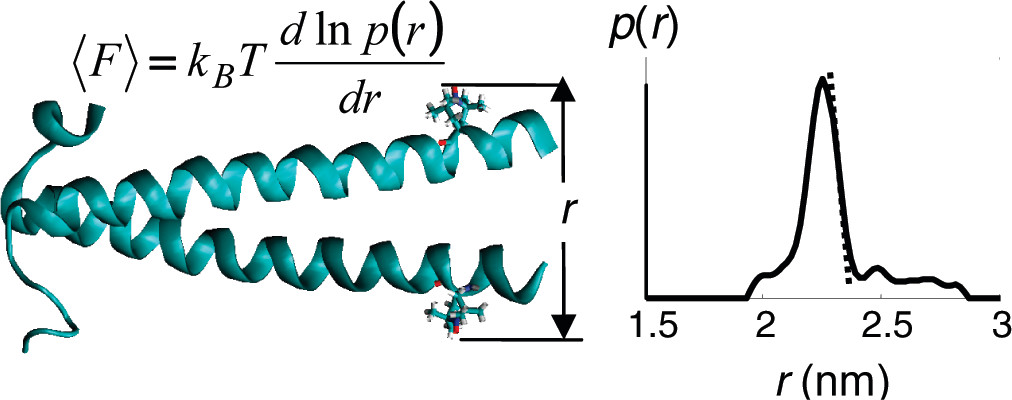
ABSTRACT: A new method for measuring forces between small protein domains based on double electron−electron resonance (DEER) spectroscopy is demonstrated using a model peptide derived from the α-helical coiled-coil leucine zipper of yeast transcriptional activator GCN4. The equilibrium distribution of distances between two nitroxide spin labels rigidly attached to the helices of the dimer was determined by DEER and yielded a closing force of 100 ± 10 pN between monomers, in excellent agreement with theoretical predictions.
ABSTRACT: Multifrequency electron spin resonance (ESR) spectra provide a wealth of structural and dynamic information about the local environment of the spin label and, indirectly, about the spin-labeled protein. Relating the features of the observed spectra to the underlying molecular motions and interactions is, however, challenging. To make progress toward a rigorous interpretation of ESR spectra, we perform extensive molecular dynamics (MD) simulations of fully solvated T4 Lysozyme, labeled with the spin label MTSSL at positions 72 and 131. These two sites have been the object of numerous experimental studies and are generally considered as prototypical solvent-exposed sites on the surfaces of α-helices. To extend the time window afforded by the MD simulations, stochastic Markov models reflecting the dynamics of the spin label side chains in terms of their rotameric states are constructed from the trajectories. The calculated multifrequency ESR spectra are in very good agreement with experiment for three different magnetic field strengths without adjusting any parameters. During the trajectories, the spin labels interconvert among a fairly large number of conformations and display a propensity to form interactions with protein residues other than their nearest neighbors along the helix. The detailed picture of the spin label emerging from the MD simulations provides useful insight into the molecular origins of the available spectroscopic and crystallographic data.
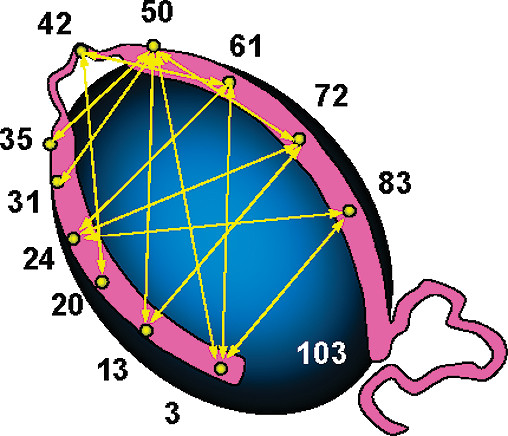
ABSTRACT: We apply pulsed dipolar ESR spectroscopy (Ku-band DEER) to elucidate the global conformation of the Parkinson's disease-associated protein, α-synuclein (αS) bound to small unilamellar phospholipid vesicles, rodlike SDS micelles, or lipid bicelles. By measuring distances as long as ˜7 nm between introduced pairs of nitroxide spin labels, we show that distances are close to the expectations for a single continuous helix in all cases studied. In particular, we find distances of 7.5 nm between sites 24 and 72; 5.5 nm between sites 24 and 61; and 2 nm between sites 35 and 50. We conclude that αS does not retain a "hairpin" structure with two antiparallel helices, as is known to occur with spheroidal micelles, in agreement with our earlier finding that the protein's geometry is determined by the surface topology rather than being constrained by the interhelix linker. While the possibility of local helix discontinuities in the structure of membrane-bound αS remains, our data are more consistent with one intact helix. Importantly, we demonstrate that bicelles produce very similar results to liposomes, while offering a major improvement in experimentally accessible distance range and resolution, and thus are an excellent lipid membrane mimetic for the purpose of pulse dipolar ESR spectroscopy.
ABSTRACT: We demonstrate the use of pulsed ESR spectroscopy to measure intramolecular distances in the Parkinson's disease-associated protein α-synuclein bound to detergent and lysophospholipid micelles. We show that the inter-helical separation between the two helices formed upon binding to micelles is dependent on micelle composition, with micelles formed from longer acyl chains leading to an increased splaying of the two helices. Our data suggest that the topology of α-synuclein is not strongly constrained by the linker region between the two helices and instead depends on the geometry of the surface to which the protein is bound.
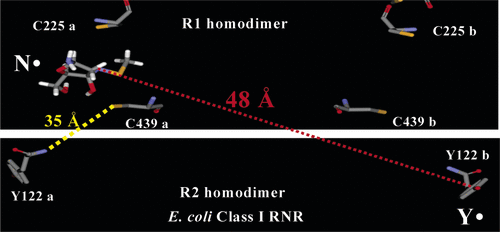
ABSTRACT: The class I E. coli ribonucleotide reductase, composed of homodimers of R1 and R2, catalyzes the conversion of nucleoside diphosphates to deoxynucleoside diphosphates. The reduction process involves the tyrosyl radical on R2 that generates a transient thiyl radical on R1 over a proposed distance of 35 Å. A mechanism-based inhibitor, 2′-azido-2′-deoxyuridine-5′-diphosphate, that reduces the tyrosyl radical on R2 and forms a nitrogen-centered radical on R1 has provided a method to measure the diagonal distance between the two subunits. PELDOR and DQC paramagnetic resonance methods give rise to a distance of 48 Å, similar to that calculated from a docking model of the R1 and R2 structures.
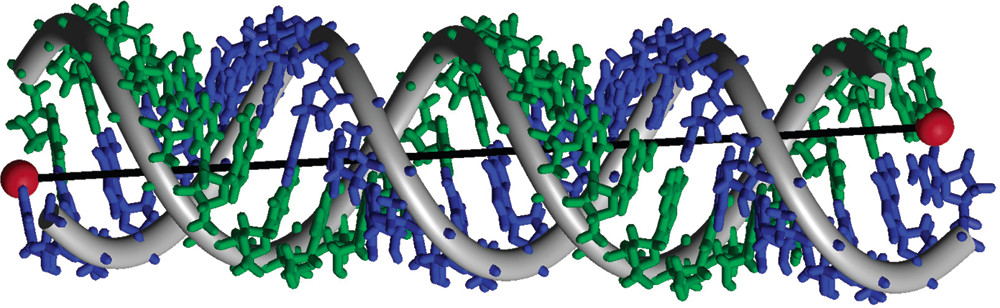
ABSTRACT: It is shown how the new technique of double-quantum filtered refocused electron spin–echoes is a significant improvement over double-quantum coherence ESR, since it increases the experimental acquisition time. This enables the measurement of longer distances in bilabeled biomolecules. The method is demonstrated on a long double-stranded A-type RNA, spin labeled at both ends. The measured distance of 72 Å is in excellent agreement with molecular modeling.
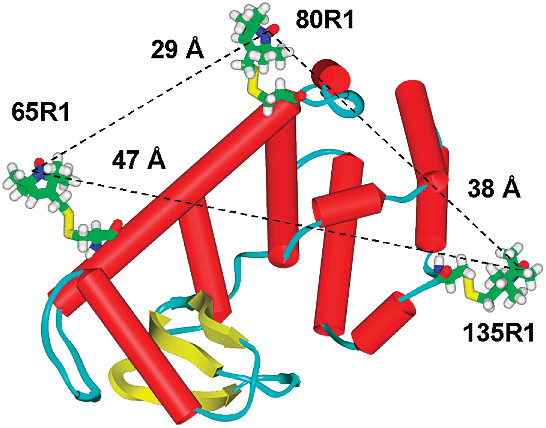
ABSTRACT: We report the use of a novel pulsed ESR technique for distance measurement, based on the detection of double quantum coherence (DQC), which yields high quality dipolar spectra, to significantly extend the range of measurable distances in proteins using nitroxide spin-labels. Eight T4 lysozyme (T4L) mutants, doubly labeled with methanethiosulfonate spin-label (MTSSL), have been studied using DQC-ESR at 9 and 17 GHz. The distances span the range from 20 Å for the 65∕76 mutant to 47 Å for the 61∕135 mutant. The high quality of the dipolar spectra also allows the determination of the distance distributions, the width of which can be used to set upper and lower bounds in future computational strategy. It is also demonstrated that the shape of these distributions can reveal the presence of multiple conformations of the spin-label, an issue of critical relevance to the structural interpretation of the distances. The distances and distributions found in this study are readily rationalized in terms of the known crystal structure, the characteristic conformers of the nitroxide side chains, and molecular modeling. This study sets the stage for the use of DQC-ESR for determining the tertiary structure of large proteins with just a small number of long-distance constraints.
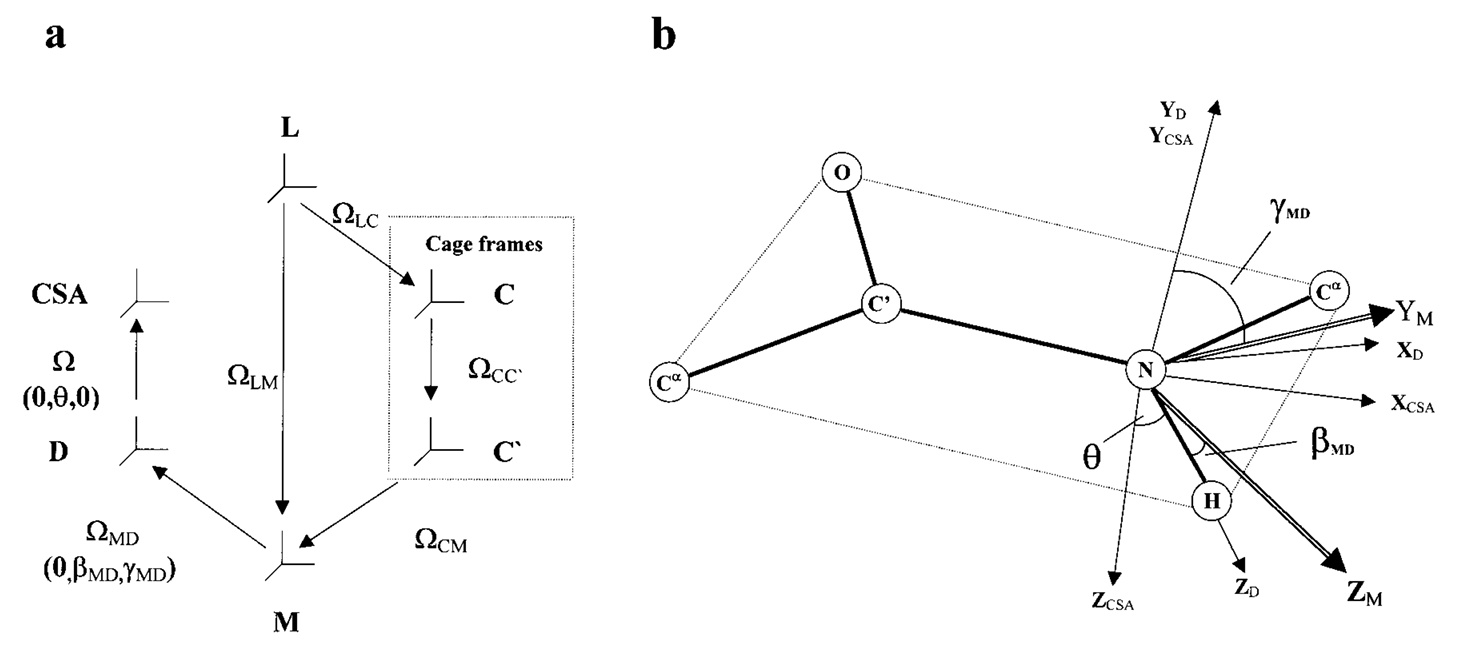
ABSTRACT: The two-body Slowly Relaxing Local Structure (SRLS) model was applied to 15N NMR spin relaxation in proteins and compared with the commonly used original and extended model-free (MF) approaches. In MF, the dynamic modes are assumed to be decoupled, local ordering at the N−H sites is represented by generalized order parameters, and internal motions are described by effective correlation times. SRLS accounts for dynamical coupling between the global diffusion of the protein and the internal motion of the N−H bond vector. The local ordering associated with the coupling potential and the internal N−H diffusion are tensors with orientations that may be tilted relative to the global diffusion and magnetic frames. SRLS generates spectral density functions that differ from the MF formulas. The MF spectral densities can be regarded as limiting cases of the SRLS spectral density. SRLS-based model-fitting and model-selection schemes similar to the currently used MF-based ones were devised, and a correspondence between analogous SRLS and model-free parameters was established. It was found that experimental NMR data are sensitive to the presence of mixed modes. Our results showed that MF can significantly overestimate order parameters and underestimate local motion correlation times in proteins. The extent of these digressions in the derived microdynamic parameters is estimated in the various parameter ranges, and correlated with the time scale separation between local and global motions. The SRLS-based analysis was tested extensively on 15N relaxation data from several isotropically tumbling proteins. The results of SRLS-based fitting are illustrated with RNase H from E. coli, a protein extensively studied previously with MF.
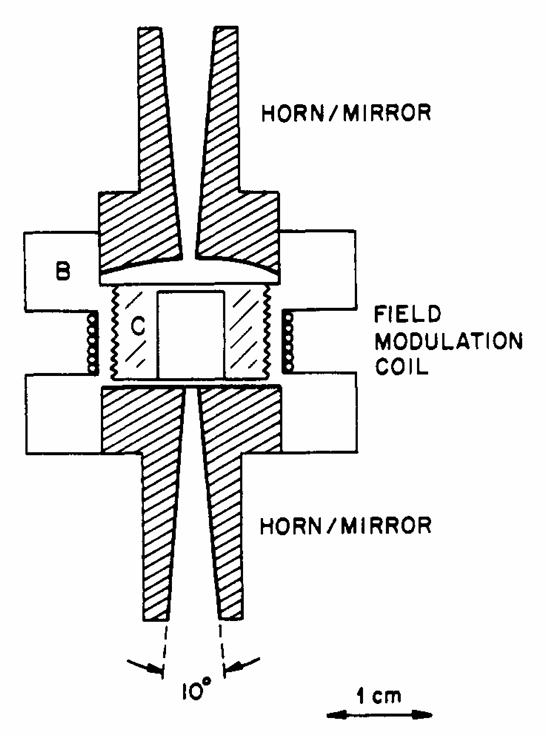
ABSTRACT: ESR spectra at 250 GHz and magnetic fields ranging from 45 to 95 kG have been measured for the compounds Mn(γ-picoline)4X2 and Mn (o-phenanthroline)2X2, where X = Cl, Br, and I. These spectra are in the high-field limit and well resolved; hence, they are inherently simple to analyze. We find these spectra to be very sensitive to the precise values of the zero-field splitting (zfs) parameters. Thus D and η (E/D) can be estimated directly from the spectrum, with very accurate values obtained using third-order perturbation theory for the electron spin Hamiltonian of high-spin Mn2+. The contributions of all five allowed electron spin transitions are observed and successfully simulated. The γ-picoline complexes have axial symmetry and D increases from 0.186 to 0.999 cm−1 with the size of the halogen. The o-phenanthroline complexes show a wide range of rhombic distortion and E increases linearly with the magnitude of D. The chloride compound is nearly axial (D = 0.124 cm−1, η = 0.04), while the iodide compound is highly distorted (D = 0.590 cm−1, η = 0.246). To demonstrate the applicability of the high-frequency ESR technique to biological samples, we have also measured the high-field spectrum of Mn(II) protoporphyrin IX and determined its zfs parameters to be D = 0.775 cm−1 and η = 0.048.
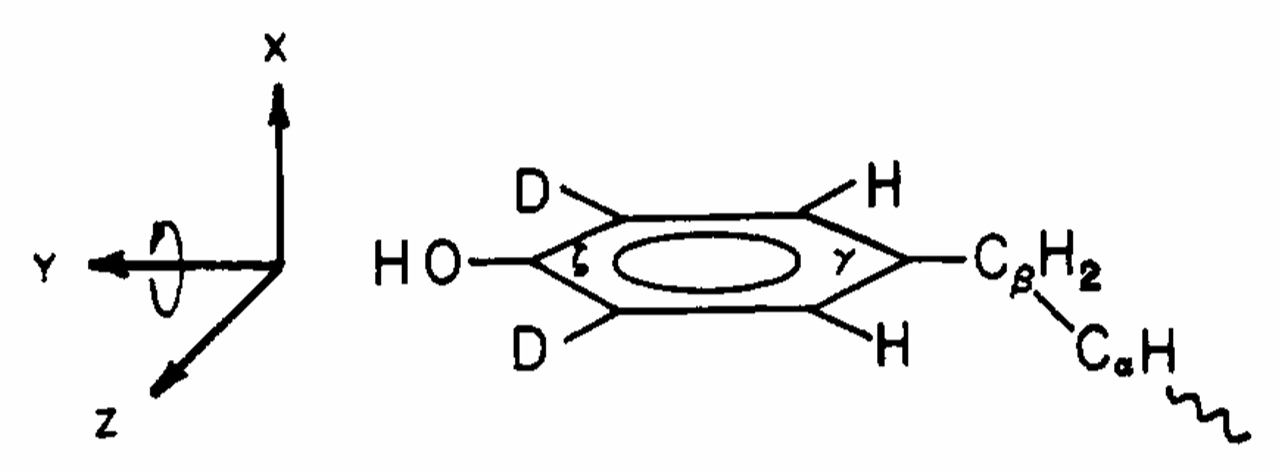
ABSTRACT: Deuterium NMR spectra of polycrystalline [tyrosine-3,5-2H2] [Leu5]enkephalin show that the aromatic tyrosyl ring of this pentapeptide is executing 180° flips about the Cβ-Cγ axis in the solid state. Specifically, the axially symmetric powder pattern observed at low temperature collapses to an axially asymmetric pattern with η ≃ 0.6 at high temperature. Computer simulations of the NMR line shapes, which account for spectral distortions induced by the quadrupole echo technique, indicate that at room temperature the flipping rate is approximately 5 × 104 s−1 and that it increases to about 106 s−1 at 101 °C.
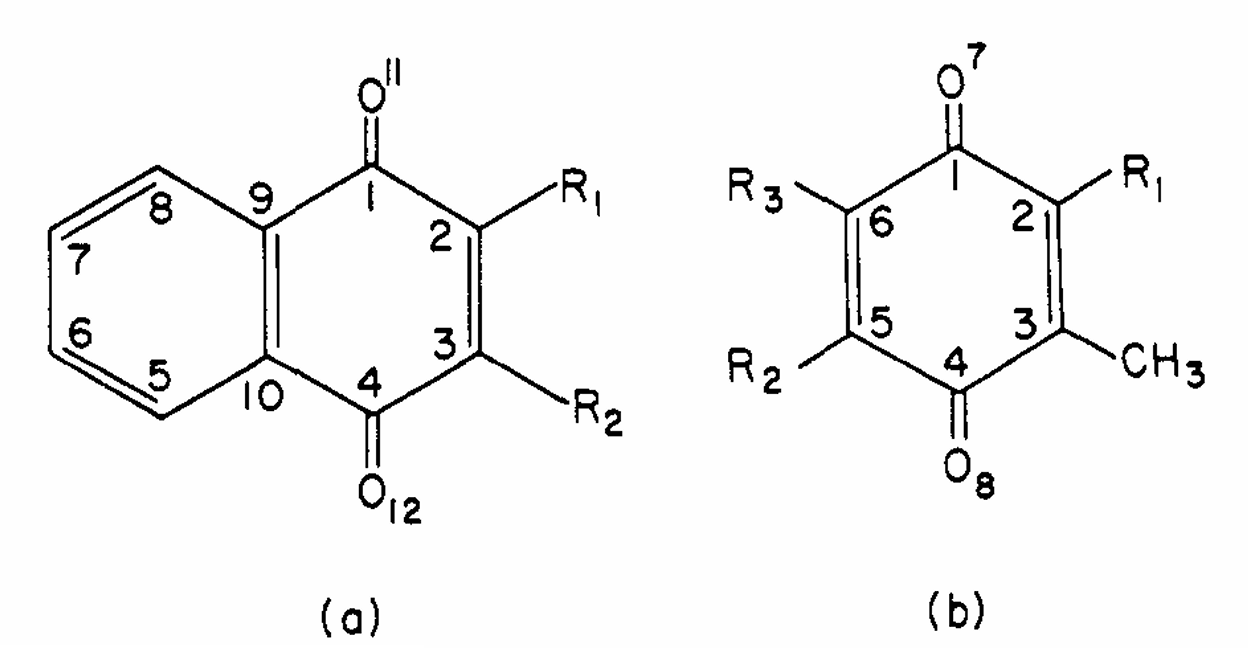
ABSTRACT: The electron nuclear double resonance (endor) spectra of ethanolic solutions of several semiquinones of biological interest have been observed, and splitting constants as well as per cent enhancement measurements have been made. The endor results, which are especially useful in obtaining unequivocal analyses of the more complex esr spectra, also showed greatly increased resolution by resolving small splitting constant differences unobservable in the esr. This latter result was due to a factor of ˜5 decrease in the endor as compared to the esr widths. These results were used to evaluate spin densities. MO calculations which were performed were found to be in satisfactory agreement with the "experimental" spin densities, including those at positions of small density. It is found that the introduction of a long side chain in the vitamin semiquinones in place of a methyl group has very little effect on the spin density distribution, including that at the ring carbon to which the side chain is attached. Alternating line width effects were observed in the esr of the semiquinones derived from vitamin K1 and coenzyme Q-10 (ubisemiquinone, USQ) with associated line width effects in the endor. They are due to rotation of the aromatic ring relative to the attached methylene protons of the side chain. At -50°, this motion is sufficiently slow in USQ in dimethoxyethane (DME) that two inequivalent methylene protons are observed in the endor and esr. Comparable activation energies and preexponential factors were found for USQ in DME and ethanol with similar results for vitamin K1.
ABSTRACT: The relaxation-matrix theory of line widths in electron spin resonance spectra has been employed to analyze the line-width effects arising from the motions of methyl groups in π-electron free radicals. Broadening of the lines can result because the methyl-proton hyperfine splittings are a function of the angle of orientation of the methyl group, and thus the splittings fluctuate as this angle varies. The motion of the methyl groups is treated by a Brownian motion model assuming a free rotatory diffusion about the C–C bond between the methyl group and the aromatic system to which it is bonded, and also by a jump model in which the group can undergo transitions from one to another of three different equilibrium orientations. Radicals with several methyl groups are analyzed as having either completely correlated or completely uncorrelated motions. The correlated motions are approximated by assuming a gear-like interleaving without slip of the hydrogen atoms on methyl groups substituted at adjacent positions on an aromatic ring. When the motions of the methyl group cause large contributions to the line width, the central pair of lines from the splittings by one methyl group are predicted to be broad and the end lines narrow. When there are two or four methyl groups in the radical, large linewidth contributions lead to a spectrum in which every third line is narrow while the remaining lines are broad, an effect which is analogous to the alternating line-width phenomenon. For very rapid rotations, nonsecular as well as secular line-width contributions are important, and consequently the nondiagonal relaxation matrix for the case of one methyl group has been analyzed in detail. None of these effects of methyl-group rotations has been observed in the e.s.r. spectra of aromatic radicals, and from the negative results it is possible to estimate that the relaxation time τc for the rotation of the methyl groups is less than 10−8 sec. The predicted effects are most likely to be found by low-temperature studies on radicals which have large methyl-proton hyperfine splittings and highly hindered methyl groups.
.svg)